


Research
Is the Brain Ready for
Physical Organic Chemistry?
a personal perspective on the potential contributions of
chemistry to neuroscience, and a summary of some recent results
Dennis A. Dougherty
Division of Chemistry and Chemical Engineering,
California Institute of Technology, Pasadena, CA 91125
[This is an substantially modified and continuously updated version of a review that appeared in the Tenth Anniversary Issue of the Journal of Physical Organic Chemistry - ref: J. Phys. Org. Chem, 11, 334-340 (1998)].
Abstract: Efforts to developed detailed insights into the structure and function of the molecules of memory, thought, and sensory perception - physical organic chemistry on the brain - are described. By combining chemical insights and methodologies with a number of techniques adapted from modern biology, it is now possible to perform systematic structure/function studies on the integral membrane proteins that play a central role in neurobiology. There are substantial challenges associated with such studies, but we believe the potential payoff is considerable.
Physical organic chemistry has always been an evolving field, receiving regular injections of revitalizing influences from new methodologies and new structural/mechanistic challenges. After the basic paradigms were established in pioneering studies, those foundations were constantly challenged and refined as new tools such as fast spectroscopy, gas phase techniques, matrix isolation, and computational chemistry produced ever more detailed insights into the nature of reactive intermediates. Now, even select transition states can be directly characterized.
Likewise, along with a vital core of issues related to the basic nature of prototype reactive structures, physical organic chemistry has always branched out to impact new areas. The influence of physical organic chemistry is easily found in organometallic chemistry, biochemistry/bioorganic chemistry, and materials science. Indeed, much of the excitement in chemistry today is generated at these "interfaces", such as chemistry/biology and chemistry/materials science, and certainly physical organic chemistry is very much involved in these challenging new directions.
Our own work has emphasized both interfaces, attempting to rationally develop organic magnetic materials,[ref] and to understand recognition phenomena such as the cation-π interaction that are relevant to many biological systems.[ref] More recently, though, our interests have moved in a new direction. From one perspective we are doing physical organic chemistry on the brain. Perhaps more correctly, we are asking whether it is possible to do so. That is why we state the title as a question - a question that just as easily could have been stated as, "Is physical organic chemistry ready for the brain?".
The issue is one of complexity. The brain is a remarkably complicated structure. Of course, implicit in the notion of thinking about the brain like a chemist, is that we are concerned with the molecules of the brain. To do physical organic chemistry on the brain is to ask structural and mechanistic questions about the molecules of thought, memory, and sensory perception. This is indeed a daunting task, but we believe new capabilities - some from chemistry and some from biology - make this a viable goal for modern physical organic chemistry. We will begin with a few comments on neuroscience in general, and then introduce the molecules of the brain. We will then describe several techniques that will be unfamiliar to most physical organic chemists but that, we feel, make it possible to think like a physical organic chemist in this arena. Historically, new methods for physical organic chemistry have come from physics (lasers, matrix isolation, …), but here it is biological tools that are being adapted to mechanistic problems. We will close with several examples from our own recent work that we would consider to be physical organic chemistry on the brain.
The Molecules of the Brain
First, a few numerical estimates concerning the brain.[ref] There are roughly 1012 neurons in a human brain, and there is great diversity among them, with perhaps 1000 different types of neurons. A "typical" neuron makes connections (synapses) with 103 - 104 other neurons - the brain is wired in a massively parallel fashion that is completely different from the design of current computers. This means there are 1015 - 1016 synapses in the human brain, involving ca. 2 million miles of axons, making the human brain the most complex structure, natural or artificial, on earth. [ref] Even the massive undertaking of completely sequencing the 3 x 109 base pairs of the human genome seems modest compared to the task of unraveling the workings of the brain.
Moving toward molecular issues, much of the action occurs at the synapse. The brain is about cell-cell communication, and the synapse is the gap between two cells - typically 30-40 nm thick. This gap is prototypically overcome by small neurotransmitter molecules that are released from the presynaptic neuron, traverse the synaptic gap, and are then recognized and processed by receptors on the surface of the postsynaptic neuron. In earlier times, efforts to apply physical organic chemistry to neurobiology focused on these small neurotransmitters - structures such as acetylcholine, dopamine, and serotonin. Now the challenge is to develop a chemical-scale understanding of the neuroproteins themselves. Here we use the term "neuroproteins" to generically identify the integral membrane proteins of the central and peripheral nervous systems, including ion channels, neurotransmitter transporters, ligand-gated and G protein-coupled neuroreceptors, and related structures. These are proteins with a tremendous diversity of structure, but a few common features.
First, since the issue is cell-cell communication, these molecules tend to be on the surfaces of cells. In molecular neurobiology surface location generally implies integral membrane proteins. That is, neuroproteins are not membrane-anchored, or structures with just one membrane-spanning segment like the family of hormone receptor molecules. Neuroproteins typically cross the membrane many times - some containing 12 or more transmembrane segments. Often more than half the protein is embedded in the membrane. It is still true that, with few exceptions, membrane embedded means not amenable to high resolution structural methods such as x-ray crystallography or NMR.[ref] For some systems, very useful low resolution images are available, but atomic-scale detail is not.
A further complication is that many, but not all, neuroproteins are multisubunit systems. Often 4 or 5 individual proteins - each with several transmembrane segments - combine to form the functional system. In addition, it is in general difficult to accumulate significant quantities of pure, properly folded, functional neuroproteins. This makes the task of characterization all the more difficult.
So, we have no high resolution structural data, very little material available, and only the vaguest notion of how these structures work - not exactly the ideal system for detailed, physical organic chemistry studies. Fortunately, one other common feature provides some rescue. The currency of the brain is current - more precisely, highly regulated ionic movements across cell membranes, which themselves have a permanent transmembrane potential. Either directly or indirectly, most of the important molecules of neurobiology are involved in the production of an ionic current. If there is one thing we can do, it is measure small electrical currents, and for some time modern electrophysiological methods have enabled the measurement of the electrical activity of a single cell. With the remarkable capabilities of the patch clamp, it is commonplace to monitor in real time the chemical activities of a single ion channel molecule! This connection to electrical activity is the first key to being able to do physical organic chemistry on the molecules of neurobiology.
Molecular Neurobiology
The staple of physical organic chemistry has been structure/function correlation. In the absence of direct observation of reactive structures and high resolution structural data, important, fundamental insights into mechanistic organic chemistry have been gained from structure/function studies. For some time, an outstanding group of neurobiologists has been performing important structure/function studies on neuroproteins. To do so, one needs two tools: a way to rationally modify the structures of the neuroreceptors; and a means to evaluate the functional consequences of the structural change. We will summarize here the now standard way in which this is done in molecular neurobiology.
The analog to organic synthesis is site-directed mutagenesis combined with heterologous expression - the former being familiar to all, the latter perhaps not. Nowadays, it is a fairly straightforward task to modify the structures of proteins by changing the codon in a given gene to code for a different amino acid. Frankly, in our experience, anyone who can do non-trivial organic synthesis can do site-directed mutagenesis, with the latter often being much easier than the former. This allows systematic structural modification and has, of course, revolutionized chemical biology.
There remains, however, a significant hurdle due to the very nature of neuroproteins - integral membrane, often multisubunit, coupled to ionic currents. Neuroproteins are generally incompatible with conventional protein expression systems. It is only meaningful to evaluate them in the context of a living cell (in vivo), where some semblance of proper functional behavior is possible. However, most protein translation systems function in a test tube (in vitro). Fortunately, modern biology provides a solution to this problem - heterologous expression.[ref] Amazingly (from a chemist's viewpoint), if one delivers into certain types of living cells the mRNA that codes for a protein of interest, the cells will obligingly translate that message into nascent protein, and then properly fold, assemble, and transport the now intact neuroprotein to the cell surface. A typical vehicle, and the one we have emphasized, is an oocyte cell from the frog Xenopus laevis. It is a large (~1mm diameter), very cooperative cell that can be used to express a wide variety of neuroproteins. Note that the mRNA injected into the Xenopus oocyte need not be from a frog gene; it can be from a human or mouse or almost any other species, hence the term heterologous expression.
Site-directed mutagenesis in combination with heterologous expression solves the "organic synthesis" problem, and electrophysiology provides the "spectroscopic tool". With these two tools and many related approaches, systematic structure/function studies of neuroproteins are feasible.
Chemical Neurobiology
The in vivo nonsense suppression method for unnatural amino acid incorporation
The capabilities of modern molecular neurobiology are impressive, but we need one more tool to do "real" physical organic chemistry. A severe limitation of site-directed mutagenesis is that it is restricted to the 20 natural amino acids. Obviously, Nature has done quite well with this set of options, but to a physical organic chemist, the structural variation presented by the natural amino acids is quite limited. For example, suppose one identifies a crucial tyrosine in a protein of interest, and one wants to know what role this residue plays. One obvious mutation is to phenylalanine, converting a phenol (Tyr) to a simple benzene (Phe). However, this causes many changes - a hydrogen bonding group is removed; the electronic nature of the aromatic ring is altered; and a significant "steric hole" is left behind. Clearly, something more subtle would be valuable. For example, O-methyl tyrosine (anisole instead of phenol) addresses the hydrogen bonding issue without seriously altering the electronic structure of the aromatic. This is perhaps the first thing a chemist would do, but the last thing a biologist would do, because O-methyl tyrosine is not a naturally occurring amino acid.
Fortunately, there is a way around this limitation. Building on the extensive biology of "suppressor" tRNAs, and some important efforts by Hecht, Peter Schultz developed, in 1989, a general method for biosynthetic incorporation of unnatural amino acids into proteins.[ref] Briefly, one uses site-directed mutagenesis to incorporate a "stop" codon at the site of interest, and a special tRNA that recognizes the stop codon (termed a "suppressor" tRNA) to incorporate unnatural amino acids at the site. This method has been applied brilliantly by the Schultz group to a number of systems using in vitro protein synthesis. Important studies on unnatural amino acid incorporation have also been carried out by the Chamberlin group.[ref]
At Caltech, in collaboration with my colleague in biology Henry Lester, we have adapted this protocol to the heterologous expression system of the Xenopus oocyte.[ref] It can be appreciated that a living cell is much different from a test tube, so this initially presented a significant challenge. However, the in vivo nonsense suppression method for unnatural amino acid incorporation is now a general methodology. As such, we now have the ability to incorporate almost any functional group into almost any location in a wide range of neuroproteins. It is this advance that we feel makes physical organic chemistry on the brain a plausible goal. Further details on the in vivo nonsense suppression methodology , including a pictorial summary of the protocol, are available. In summary, we can incorporate a wide range of unnatural amino acids into all the major types of integral membrane proteins involved in synaptic transmission.
{kind=link}
This new capability opens up an essentially limitless number of experiments designed to probe structure/function issues in neuroproteins. We can now do chemical biology on the molecules of the nervous system - what we term chemical neurobiology. Over the past few years, a major focus of the Lester/Dougherty collaboration has been to develop an appreciation of what types of experiments are best suited to this protocol. On reflection, one can imagine two different types of applications: incorporating structural variations that are more subtle than is possible with the natural amino acid set, allowing systematic structure/function studies; or incorporating unnatural amino acids that are wildly different from Nature's set, allowing totally new structure/function probes. We have pursued both, and will briefly outline examples of each.
Physical organic chemistry on the nicotinic receptor
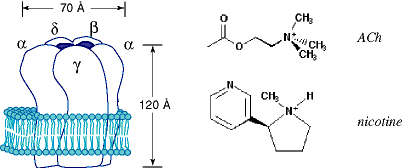
Although the unnatural amino acid methodology is applicable to a wide range of neuroproteins, we will use just one system to illustrate our efforts at physical organic chemistry on the brain. The nicotinic acetylcholine receptor (nAChR) is the major neuroreceptor at neuromuscular junctions, and is increasingly recognized to be important in the brain (Figure 1).[ref] It is the prototypical ligand-gated ion channel. When agonist (ACh) is released into the synapse by a presynaptic neuron, it binds to postsynaptic nAChRs, causing a cation-specific ion channel contained within the receptor to open. Nicotine is a competitive agonist of the nAChR, and certainly the neuronal nAChRs play an important role in nicotine addiction. The nAChR is also the best studied, prototypical member of a large class of neuroreceptors, that also includes receptors for GABA, glycine, and serotonin. The nAChR (see below) is a pentamer formed from four homologous subunits, a, b, g, d, in a ratio a2bgd. The agonist binding site is thought to be primarily associated with the a subunit, and so there are two agonist binding sites that interact in a positively cooperative way. We emphasize that it is not our goal here to provide a comprehensive discussion of the nAChR. Rather, we hope to provide a sense of the kinds of chemistry that are now possible with neuroproteins, and we will use the nAChR as a vehicle for that discussion. For those interested in further details, a number of excellent reviews emphasizing structural issues in the nAChR are available.[ref]
Classical structure/function studies.
Identifying the cation-π binding site of the nicotinic receptor. The cation-π interaction - the highly stabilizing interaction between a cation and the face of an aromatic ring - has been a major research focus of our group for some time. We have established the generality of this phenomenon in gas phase interactions, artificial receptors in aqueous media, and in biological receptors.[ref] In particular, we proposed some time ago that binding sites for acetylcholine would be especially likely to use cation-π interactions to bind the quaternary ammonium group of ACh, a prediction that was subsequently validated by the crystal structure of the enzyme acetylcholine esterase.
A large number of aromatic residues have been identified as being near to and/or contributing to the ACh binding site of the nAChR. However, proximity does not prove a cation-π interaction. The question is, how does one recognize a cation-π site in a large protein? We have introduced a wide range of systematic structural variations at several aromatic sites, hoping to identify one or more as cation-π sites. Initial studies focused on tyrosines.[ref] These produced several interesting results, but no clear cation-π sites. Subsequent theoretical studies indicated that tryptophans are the best of the natural amino acids for cation-π interactions. In addition, we have established that computational models can accurately predict the cation-π binding ability of an aromatic ring. Using this information, we have more recently used the suppression methodology to incorporate a wide variety of tryptophan derivatives at several putative cation-π sites of the nAChR. At one and only one site - Trp149 of the alpha subunit - did we see a strong effect.[ref]
Shown below is a plot that documents this effect. On the y-axis is the EC(50) for ACh at the receptor for Trp and the four fluorinated derivatives shown. EC(50) is not technically a binding constant for ACh, but in this context it is highly likely that variations in EC(50) are tracking variations in ACh affinity for its binding site. On the x-axis is the "cation-π binding ability" of the ring. This number is obtained from an ab initio, quantum mechanical calculation on a gas phase, small molecule, model system (Na+ binding to the relevant indole). Thus, on one axis we have the results of an a priori, quantum mechanical calculation, and on the other axis a measurement on an intact, MW = 290,000, ion channel protein in a living cell - a huge range of scale. Yet, the correlation is undeniable.
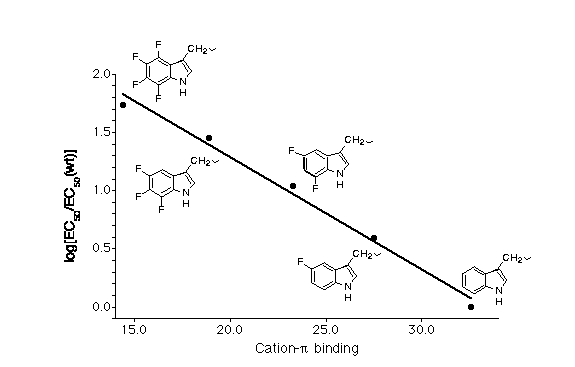
We feel this data unambiguously establishes that Trp149 of the alpha subunit is the cation-π binding site. In particular, that data indicate that the quaternary ammonium group of ACh makes van der Waals contact with the six-membered ring of the indole sidechain. This is the highest precision structural information to date on a neuroreceptor, and it establishes the power of the unnatural amino acid mutagenesis strategy.
Hydrophobic residues in the ion channel. Another region where systematic structure/function studies have proved valuable is in the actual ion channel part of the receptor - the pore through with cations flow once the channel is placed into the open state as a consequence of agonist binding. In particular, a crucial leucine residue - the so-called 9' residue - in this region is thought to play an important role in the "gating" of the ion channel.[ref] In one model, five leucines - one from each subunit - are proposed to form a "hydrophobic plug" in the closed state, with channel opening involving a swinging of the Leu sidechains away from the pore. Using the in vivo nonsense suppression method, we were able to test this hypothesis by systematically altering the hydrophobicity of the 9' sidechain.[ref] Shown below are typical comparisons that are possible with the unnatural amino acid methodology. We had a number of ways of studying homologous series - adding one CH2 group - to increase hydrophobicity. More subtle were the comparisons such as O-Me-threonine vs. isoleucine, in which the sterics were almost identical but the hydrophobicities were significantly different.
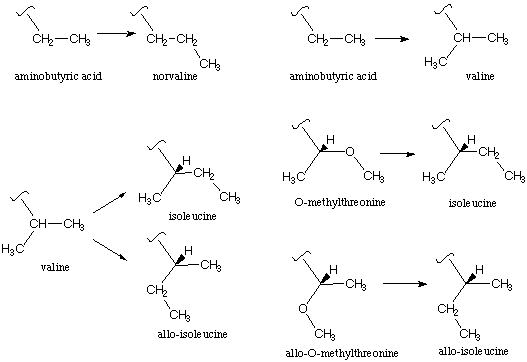
In most comparisons, increased hydrophobicity at the 9' position did make the channel more difficult to open, consistent with the hydrophobic plug model. However, at one site, the 9' position of the d subunit, a very subtle effect was seen. At this site only, the stereoisomeric sidechains of isoleucine (Ile) and allo-isoleucine (aIle) gave measurably different responses. This is clearly not a hydrophobic effect, and it suggests that a more subtle, highly structured feature is involved. Further analysis of this result led to the proposal of a special pair relationship between the b and d subunits and, perhaps, a revision of the proposed gating model.
It is worth contemplating the Ile/aIle comparison a bit. The nAChR is a large protein with a MW of ca. 290,000, five subunits, and 20 membrane spanning segments. Yet two diastereomeric receptors differing only in the relative positions of methyl vs. ethyl groups can be distinguished easily, a distinction often not possible in small molecules. This attests to the power of electrophysiology, and augurs well for systematic structure/function studies of neuroproteins.
More Dramatic Mutations
We have also developed unnatural amino acids that are far outside the realm of the natural set, and we will briefly outline some examples here.
SNIPP. Illustrated below are the unnatural amino acid (2-nitrophenyl)glycine (Npg) and the expected results from irradiation of a protein containing this novel residue. When incorporated into a protein, Npg gives rise to an o-nitrobenzyl amide group. The o-nitrobenzyl group has been extensively used as a photochemically removable protecting group for alcohols, amines, carboxylates, amides and related heteroatom functionalities.[ref] In the present context, following through the usual o-nitrobenzyl photochemistry with Npg leads to cleavage of the peptide backbone. Such a site-specific, nitrobenzyl-induced, photochemical proteolysis (SNIPP) could be quite useful in evaluating which domains of a complex protein are crucial to specific functions, and which are less so.
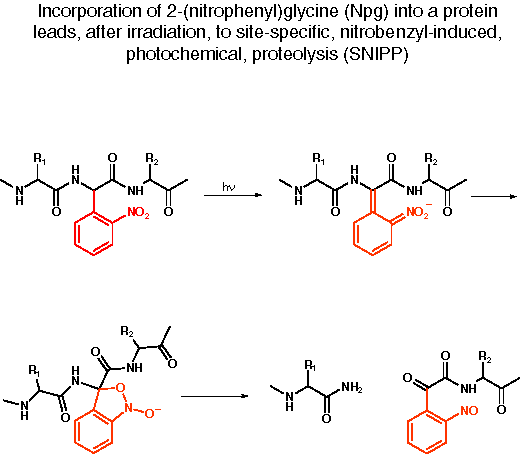
In a number a recent experiments involving the nAChR and other ion channel proteins, we have established that irradiation of an intact Xenopus oocyte expressing a protein containing Npg site-specifically incorporated by the in vivo nonsense suppression method does lead to cleavage of the protein backbone.[ref] We have used SNIPP to evaluate several crucial structural features of the nAChR, and we anticipate extensive application of this methodology to a wide array of neuroproteins.
"Caged" Sidechains. A related bit of photochemistry, and one that has been used extensively in other contexts, is the use of so-called "caged" residues. This approach is well-suited to the unnatural amino acid methodology, and we have used a Tyr with the OH protected (caged) as a photoremovable o-nitrobenzyl ether (Tyr-ONB). This more conventional type of photochemistry has also produced interesting results. In particular, incorporation of Tyr-ONB at residues near the agonist binding site that are normally tyrosine produces receptors that are unresponsive to ACh. However, photolysis rescues the receptor, and restores wild type function.[ref]
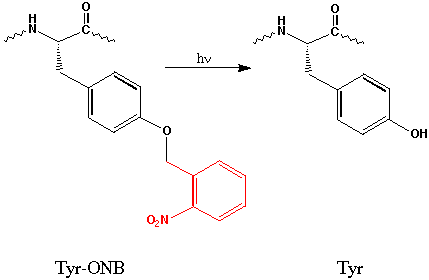
The caged tyrosine approach has also been amenable to time resolved studies. Using a modified electrophysiology rig that focuses the output of a flash lamp on an oocyte that is already "wired" for electrophysiological recording, we can record responses in the msec time domain. Interestingly, we see both a fast (t ca. 10 msec) phase and a much slower (t = 50 - 2000 msec) phase, with the latter differing considerably between the two sites of incorporation of Tyr-ONB. The implications of these findings for nAChR behavior are still under investigation, but already the potential usefulness of incorporating caged residues into neuroproteins is evident.
Expanding this approach to caged serine and threonine should be straightforward. Given the universal importance of Ser/Thr kinases and Tyr kinases in a diverse range of signalling pathways - including perhaps those involved in learning and memory - we feel the caged sidechain approach has unlimited potential.
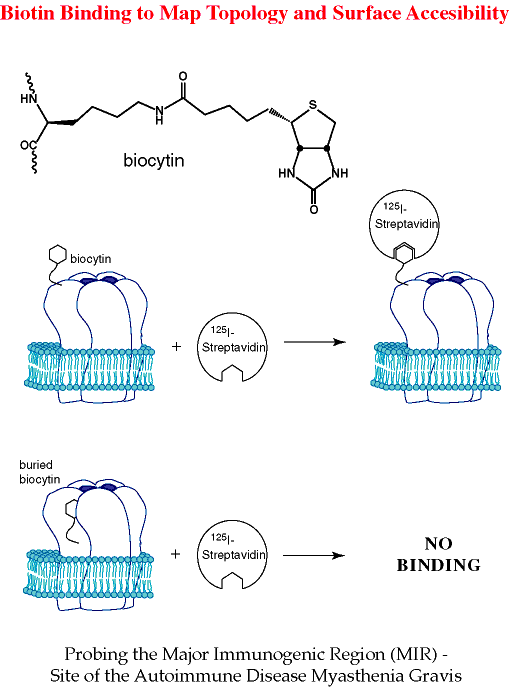
Biocytin. Another interesting, "highly unnatural" amino acid we have incorporated into a number of sites in the nAChR is biocytin. Despite its relatively large size, biocytin is very compatible with the suppression methodology. Of course, the value of biocytin as an unnatural amino acid is that it allows the exploitation of the much used biotin/streptavidin system.[ref]
A common issue in studies of complex neuroproteins is the transmembrane "topology" of the protein. In the neuroprotein field, topology has a different meaning than in conventional chemistry. Topology refers to the "ins" and "outs" of a transmembrane protein - whether the N and C termini are on the inside or the outside of the cell, and how many times the chain traverses the membrane. When no direct structural information is available, this simple issue can be difficult to resolve. Although a number of approaches to topology mapping exist, all have their own advantages and disadvantages, the latter mostly stemming from the fact that major structural perturbations are introduced, making the ultimately deduced topology suspect.
We envisioned biocytin as a relatively non-perturbing tool for evaluating transmembrane topology. As summarized in the graphic below, if biocytin is incorporated into a surface exposed residue, treatment of the oocyte with 125I-labeled streptavidin should irreversibly label the cells in an easily detectable manner. If instead the biocytin is intracellularly located, or extracellular but buried, it will not be accessible to streptavidin, and no labeling should occur. Again using the nAChR as a testing ground, we have verified the basic validity of this concept. Using a region of the receptor known to be surface exposed, we were able to label biocytin-containing receptors with streptavidin.[ref] The structural requirements for this interaction appear to be fairly stringent, as only the most highly exposed residues could be complexed. This is thus a more demanding test than just topology - the biocytin approach evaluates surface exposure (a residue can be topologically "out" but still buried). We anticipate many uses for biocytin in evaluating surface exposure.
Conclusions
There is no doubt that the molecules of molecular neurobiology are extremely complex, and that they operate in an even more complicated environment. Chemists should also appreciate that the totally reductionist view that a detailed understanding of these molecules will be equivalent to a detailed understanding of the brain is certainly wrong. Much of how the brain functions depends on "context" - the organizational and communication issues at all levels - molecule-to-molecule; cell-to-cell; region-to-region of the brain. Nevertheless, it is certainly true that a detailed understanding of the brain will require a detailed understanding of molecular neurobiology. This is especially true when considering pharmaceutical design or efforts to understand the complex effects of a wide range small molecules - both helpful and harmful - on brain function.
We have argued here that physical organic chemistry is ready to tackle the challenge of molecular neurobiology. Substantial challenges remain. It can be disconcerting to attempt mechanistic studies on a system where you know from the outset that you will never isolate pure samples of substrate or product; never get an NMR or an IR; and you are operating in an extremely complex, dynamic, heterogeneous, "impure" environment (a living cell). In addition, the recent advent of high resolution crystal structures promises to have a major impact on the field.[ref] We believe there are tremendous opportunities for physical organic chemists to have a significant impact on an undeniably important area of science.
Acknowledgment. Much of our work in molecular neurobiology results from a fruitful and highly enjoyable collaboration with Professor Henry Lester. I thank the many excellent graduate students, postdocs, and undergraduates who have contributed to these projects. Our work at Caltech has been supported by the NIH (NS 34407).
References
D. A. Dougherty, Acc. Chem. Res. 23, 88-94 (1991) - a summary of our early work on high spin molecules.
Lead references to the cation-π interaction: D. A. Dougherty and D. A. Stauffer, Science 250, 1558-1560 (1990); D. A. Dougherty, Science 271, 163-168 (1996); J. C. Ma and D. A. Dougherty, Chem. Rev. 97, 1303-1324 (1997).
Excellent introductions to molecular neurobiology: An Introduction to Molecular Neurobiology; Z. W. Hall, Ed.; Sinauer Associates, Inc.: Sunderland, MA, 1992, pp 555.; C. U. M. Smith Elements of Molecular Neurobiology; John Wiley & Sons: New York, 1989.; B. Hille Ionic Channels of Excitable Membranes; Sinauer Associates, Inc.: Sunderland, MA, 1992.
Membrane Protein Structure; S. H. White, Ed.; Oxford University Press, Inc: New York, 1994, pp 380.
Recently, the first two crystal structures of ion channel proteins have appeared.
A 3.2A crystal structure of a bacterial K+ channel has been reported by the MacKinnon group at Rockefeller Univerity: MacKinnon and coworkers, Science 280, 69-77 (1998). For a commentary from the chemists' perspective, see: D. A. Dougherty and H. A. Lester, Angewandte Chemie, International Edition, 37, 2329-31 (1998).
The Rees group at Caltech has reported a structure of a bacterial mechanosensitive channel: Rees and coworkers, Science 282, 2220-2226 (1998)
These results promise to revolutionize the ion channel field, and future work from our labs will make use of this invaluable structural information.
H. A. Lester, Science 241, 1057-1063 (1988).
C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science 244, 182-188 (1989); V. W. Cornish, D. Mendel and P. G. Schultz, Angew. Chem. Int. Ed. Engl. 34, 621-633 (1995); J. S. Thorson, E. Chapman and P. G. Schultz, J. Am. Chem. Soc. 117, 9361-9362 (1995); J. S. Thorson, E. Chapman, E. C. Murphy, P. G. Schultz and J. K. Judice, J. Am. Chem. Soc. 117, 1157-1158 (1995).
J. D. Bain, C. G. Glabe, T. A. Dix, A. R. Chamberlin, J. Am. Chem. Soc. 111 8013-8014 (1989); L. E. Steward, C. S. Collins, M. A. Gilmore, J. E. Carlson, J. B. A. Ross, A. R. Chamberlin, J. Am. Chem. Soc. 119 6-11 (1997).
Development of the in vivo suppression methodology, and early studies of the nAChR agonist binding site: M. W. Nowak, P. C. Kearney, J. R. Sampson, M. E. Saks, C. G. Labarca, S. K. Silverman, W. Zhong, J. Thorson, J. N. Abelson, N. Davidson, P. G. Schultz, D. A. Dougherty and H. A. Lester, Science 268, 439-442 (1995); M. E. Saks, J. R. Sampson, M. W. Nowak, P. C. Kearney, F. Du, J. N. Abelson, H. A. Lester and D. A. Dougherty, J. Biol. Chem. 271, 23169-23175 (1996); P. C. Kearney, N. W. Nowak, W. Zhong, S. Silverman, K., H. A. Lester and D. A. Dougherty, Mol. Pharmacol. 50, 1401-1412 (1996)
A study of the M2 Leu9' resdiue: P. C. Kearney, H. Zhang, W. Zhong, D. A. Dougherty and H. A. Lester, Neuron 17, 1221-1229 (1996).
Key references on the Nicotinic Receptor: A. Karlin, Curr. Opin. Neurobiol. 3, 299-309 (1993); A. Devillers-Thiéry, J. L. Galzi, J. L. Eiselé, S. Bertrand, D. Bertrand and J. P. Changeux, J. Membrane Biol. 136, 97-112 (1993); H. A. Lester, Annu. Rev. Biophys. Biomol. Struct. 21, 267-292 (1992); D. A. Dougherty, Pure Appl. Chem. 69, 1969-1977 (1997); N. Unwin, J. Mol. Biol. 229, 1101-1124 (1993); N. Unwin, Nature 373, 37-43 (1995); J. Machold, C. Weise, Y. Utkin, V. Tsetlin and F. Hucho, Eur. J. Biochem. 234, 427-430 (1995).
From Ab Initio Quantum Mechanics to Molecular Neurobiology: A Cation-â„– Binding Site in the Nicotinic Receptor. W. Zhong, J. P. Gallivan, Y. Zhang, L. Li, H. A. Lester, and D. A. Dougherty, Proc. Natl. Acad. Sci. (USA), 95, 12088-12093 (1998)
J. A. McCray and D. R. Trentham, Annu. Rev. Biophys. Chem. 18, 239-270 (1989).
L.-L. Galzi, A. Devillers-Thiéry, N. Hussy, S. Bertrand, J.-P. Changeux and D. Bertrand, Nature 359, 500-505 (1992); C. Labarca, M. W. Nowak, H. Zhang, L. Tang, P. Deshpande and H. A. Lester, Nature 376, 514-516 (1995); G. N. Filatov and M. M. White, Molec. Pharm. 48, 379-384 (1995).
P. M. England, H. A. Lester, N. Davidson and D. A. Dougherty, Proc. Natl. Acad. Sci. USA 94, 11025-11030 (1997).
J. Miller, S. K. Silverman, P. M. England, H. A. Lester, and D. A. Dougherty, Neuron, 20, 619-624 (1998).
N. M. Green, Advan. Protein Chem. 29, 85-133 (1975); Avidin-Biotin Technology; M. Wilchek and E. A. Bayer, Eds.; Academic Press, Inc.: San Diego, CA, 1990; Vol. 184, pp 746; K. Hofmann, G. Titus, J. A. Montibeller and F. M. Finn, Biochemistry 21, 978-984 (1982).
J. P. Gallivan, H. A. Lester, and D. A. Dougherty, Chemistry and Biology, 4, 739-749 (1997).