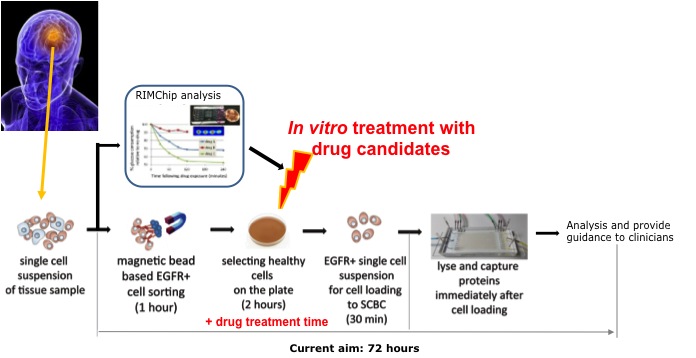
Metastatic melanoma is difficult to treat with conventional cancer therapies but many promising immune-related treatments are being developed. One such treatment is adoptive cell transfer (ACT) therapy, in which the patient’s own T cells are harvested and targeted against the tumor. However, some patients show little response, whereas others show partial to complete responses. One major observation from previous ACT trials was significant changes in T cell cytokine secretion.
We are using the levels of cytokines secreted from T cells in a predictive model for therapy response. Our current model uses T cell polyfunctionality to predict patient response to ACT using a unique “Polyfunctional Index” parameter.

The interactions of individual cells would have influence on the development of the system’s architecture. But such an influence is not clear to date. Here we are specifically interested in the influence of tumor cell interactions on the tumor architecture, i.e., phenotype spatial-temporal distribution.
We combine theory and experiment to investigate how growth-factor driven protein signaling depends upon the distance separating pairs of cancer cells. We developed a thermodynamic-derived theory to identify the intercellular separation that corresponds to the steady state of protein signaling, and validated this approach in bulk culture.
Functional proteins such as cytokines and growth factors represents the function of a cell. How the underlying transcriptional networks regulate functional protein expression is not well understood, given the cellular heterogeneity of many biological systems.
We accordingly developed a technology to quantify multiple proteins and mRNAs from the same single cells. With this technology, we are interested to address some important, fundamental biomedical questions: why only a small portion of T cells are polyfunctional, how embryonic stem cells are supporting growth to each other, and why part of hematopoietic stem cells directly functions to promote inflammation.
Blake Farrow
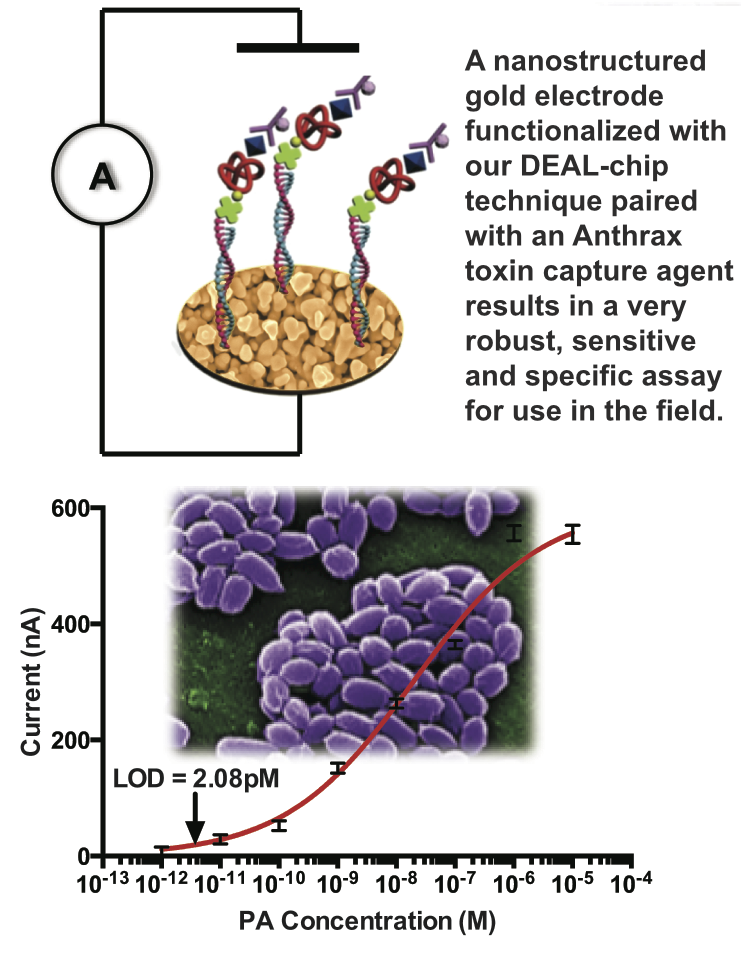
The stability and engineer-ability of protein-catalyzed click capture agents allows the creation of small, easily synthesized, and robust peptide-based macromolecules with extremely specific binding to not only a protein target of interest, but to a specific functional location on a target protein. This technology allows exquisite control over the nature and specificity of the protein-capture event, allowing application not only in standard biodetection assays, but also in much more complex milieus and applications including in vivo imaging and inhibition.
Various defense departments (including the Army Research Lab and the Defense Threat Reduction Agency) are interested in replacing monoclonal antibodies against military targets in the so-called Critical Reagents Program. These protein capture agents could be fielded in robust assays to detect toxins such as B. anthracis (Farrow et al, ACS Nano, 2013), or as designer anti-toxins engineered to be more effective and have a much broader therapeutic window than traditional antisera treatments for bio-terror agents such as botulinum neurotoxin or smallpox.
Kaycie Deyle, Samir Das, Arundhati Nag, Ryan Henning
Peptide affinity agents that mimic the performance of antibodies but maintain the stability of short peptides can be created through the use of iterative in situ click chemistry. We have used this technology to develop a plethora of capture agents that bind to target oncoproteins. Originally designed as cheaper, more efficient detection agents, these capture agents have more recently been shown to penetrate cells and function as enzyme inhibitors or activators.
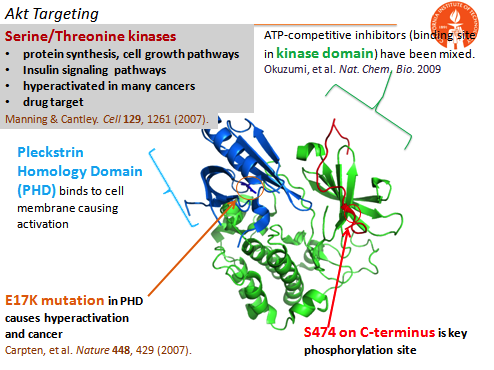
Our model oncoprotein target has been the commonly mutated enzyme Akt. Using lab-developed phospho epitope-targeting strategies, we have created capture agents that bind near the Akt Ser473 phosphorylation site, and can either inhibit or activate Akt2 in cells without targeting Akt1 or Akt3. A similar epitope-targeting strategy has also allowed for the single amino acid point mutation targeting of the E17K mutation in the Pleckstrin Homology Domain. This capture agent can inhibit the mutant protein selectively over the wildtype protein. Previous work in the lab has also provided an allosteric inhibitor of Akt1.
Current research in this area is focused on exploring the full extent of our protein manipulation capabilities, as well as targeting other common intracellular oncoproteins. The use of capture agents as cancer drugs can not only improve upon the current antibody-based therapies, but can greatly expand the range of targets due to the cell-penetrating ability of our ligands.
JingXin Liang, Arundhati Nag, Samir Das, Aiko Umeda
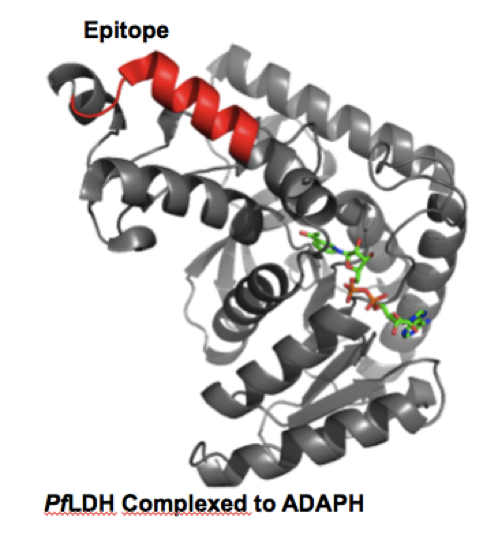
Malaria is a global health crisis caused by the parasite plasmodium. The most fatal species, plasmodium falciparum, kills over 600,000 people per year. Proper treatment measures for disease eradication rely on rapid and accurate diagnoses of the disease. Current rapid diagnostic tests (RDTs) in the field rely on antibodies which are not ideal due to their thermal instability and susceptibility to biochemical processes.
We are developing multiligand capture agents as antibody replacements in RDTS. The current focus is on both pan-species and falciparum specific disease biomarkers. A sandwich pair of capture and detection agents has been developed that exhibits high affinity and selectivity for the plasmodium falciparum lactate dehydrogenase antigen. Capture agent development against other antigens is ongoing.
The Heath group is always looking for talented and passionate scientists interested in investigating the interface between chemical physics and biology.
Email us!